
The most reliable recipes in density functional theory were never meant for ingredients that refuse to pick a direction. For decades, the standard “collinear” functionals — the workhorses that calculate the properties of everything from silicon chips to steel alloys — assumed that every electron’s magnetic moment simply pointed up or down, like a battalion of needles all aligned along the same magnetic field. The assumption was reasonable: in an ordinary ferromagnet, that’s precisely what happens. But nature loves to twist. In materials where spin‑orbit coupling is strong or where the lattice geometry frustrates alignment, magnetic moments fan out into spirals, vortices, or completely disordered patterns. The old recipes fell silent. To speak the language of these “noncollinear” magnets, theorists patched together heuristic corrections or built entirely separate functionals from scratch. What was missing — and what a new preprint by Konstantin Gaul at the Helmholtz Institut Mainz (arXiv:2605.31203) now provides — is a first-principles demonstration that the two worlds are not separate at all. With a single, universal rotation, the collinear kitchen can serve noncollinear dishes.
Think of the problem this way. In a collinear picture, every site in the material has its magnetization density pointing either along (+z) or (-z). The exchange‑correlation energy that makes density functional theory tick depends on this density and, for the most widely used semilocal functionals, on how that scalar “up‑ness” varies from place to place — its gradient. The recipes (the functionals) were built and tested exclusively for this scalar landscape. In a noncollinear magnet, the density becomes a genuine vector field. You can’t just stuff a three‑component magnetization into a functional that expects a single number; you need to know how the functional responds to longitudinal variations (those that change the magnitude of the moment) separately from transverse ones (those that tilt its direction). The practical community, most notably through the widely adopted Scalmani–Frisch scheme, found a pragmatic shortcut: they constructed a noncollinear energy expression by effectively treating the magnitude of the vector magnetization as the collinear density, but the justification was essentially a plausible guess. Rigorous underpinnings were lacking. Gaul’s work shows that the guess was correct to first order, and then systematically improves it, using a symmetry that the electrons themselves obey.
That symmetry is SU(2). In the language of spin, SU(2) is the continuous group of rotations that mixes up and down components into any arbitrary superposition. When you write the exchange‑correlation energy in a (2\times2) matrix form — what Gaul calls a (u(2)) representation — the collinear description corresponds to choosing a specific “gauge” where the matrix is diagonal. A noncollinear state, in the same representation, is non‑diagonal. But because the underlying physics is covariant under SU(2) rotations, there must exist a unitary transformation that converts the noncollinear energy into the collinear one, just as a rotation of coordinate axes doesn’t change the length of a vector. Gaul derives exactly this rotation. It’s a universal dial that, once turned, remaps any orientation of the magnetization onto a reference frame where the collinear functional can be applied directly. The trick is not merely cosmetic; the rotation introduces new gradient terms that separate the longitudinal and transverse responses, revealing that the functional’s sensitivity to tilting is genuinely different from its sensitivity to stretching.
This is not a matter of stubbornness on the part of the spins; it’s a direct mathematical consequence of how exchange and correlation energies couple to the direction of magnetization. The paper unpacks the details using a gradient expansion — a controlled approximation that organises the functional by how many derivatives of the density it contains. At the lowest, local‑density level, things are trivial: the energy depends only on the magnitude of the magnetization vector, so the rotation changes nothing. The first non‑trivial order, the generalized gradient approximation, is where the story becomes rich. Gaul shows that the functional now acquires two separate linear coefficients, one for the longitudinal part of the gradient and one for the transverse part, and that their ratio is a known function of the local polarization. The Scalmani–Frisch scheme, it turns out, corresponds to taking the first term in a systematic expansion that relates these coefficients. By keeping higher orders and performing the SU(2) rotation exactly, Gaul obtains what he calls the “NC‑SU(2)” functional — a noncollinear semilocal functional that is as well‑founded as its collinear ancestors.
You can almost picture the transformation as a kind of universal adapter. Imagine you have a collection of kitchen scales that were calibrated to weigh single‑flavour ingredients — say, only pure vanilla or pure chocolate. The scales are exquisitely precise at their one job. Then someone brings you a marbled cake batter, in which the two flavours swirl together in every imaginable proportion. The old scales are confused; their calibration makes no sense for mixtures. The SU(2) rotation is equivalent to discovering that the marbling itself can be expressed as the result of a specific stirring pattern on two pure‑flavour bases. Once you account for that stirring — once you apply the rotation — the original scales become perfectly adequate again. Of course, unlike cake batter, electron spins can exist in genuine superpositions of up and down, and the “stirring” is a coherent quantum rotation, not a classical mixing. The limits of the analogy are worth stating clearly: what gets transformed is not the physical magnetization, but the mathematical framework within which its energy is evaluated, a distinction that keeps the physics honest.
With the rigorous framework in hand, Gaul tests it on a prototypical frustrated magnet, the Cr₃ cluster. Here three chromium ions form a triangle, and antiferromagnetic exchange interactions would like to put the spins at 120° angles to one another — a classical noncollinear state. A proper noncollinear functional must be able to describe not only the energy of such a state but also the local magnetic torque, the tendency of each spin to precess under the effective exchange‑correlation field. The paper compares four levels of approximation: the lowest‑order solution based on the Kohn–Sham orbitals alone, the Scalmani–Frisch perturbative correction, a first‑order perturbative extension, and the full NC‑SU(2) rotation. The torque maps are visibly different. In the full NC‑SU(2) treatment, the exchange‑correlation magnetic field (the effective force that determines the static equilibrium orientation) aligns more naturally with the magnetization density, producing a self‑consistent picture where the torque vanishes, as it must for a stationary state. This is a stringent consistency test, and it passes.
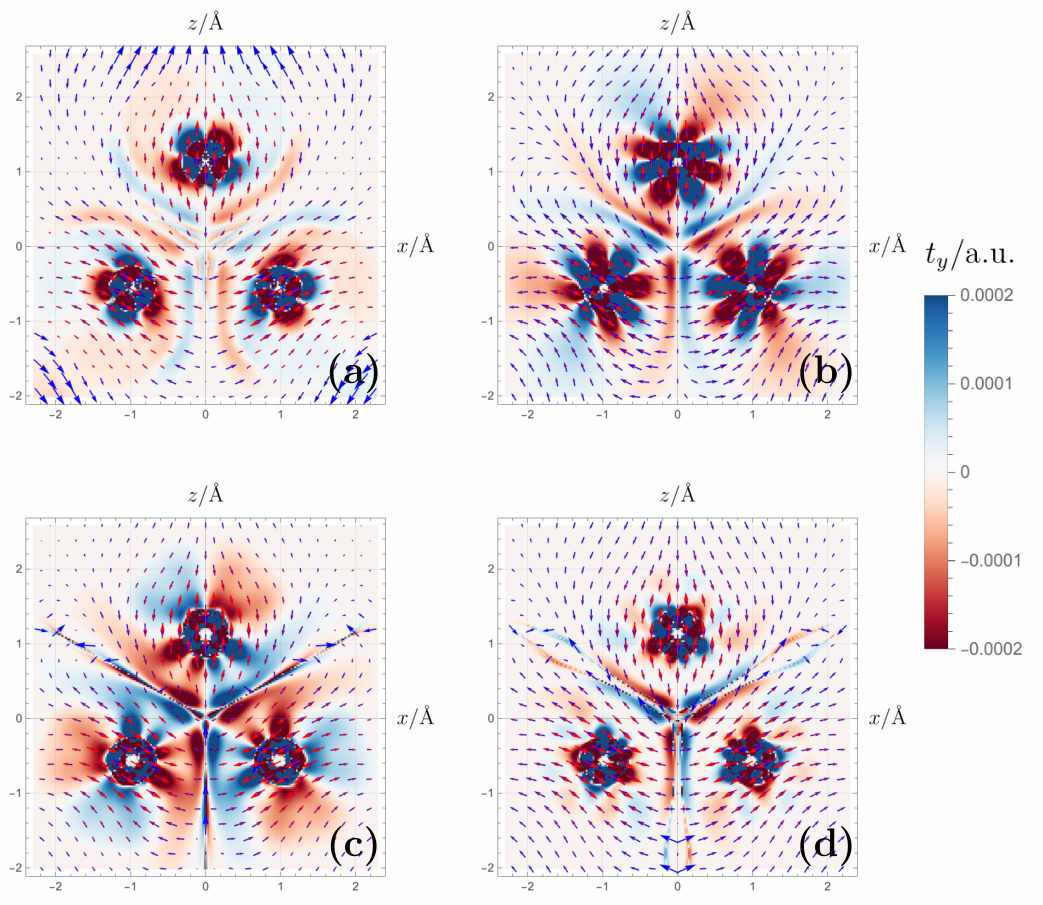
Magnetic torque patterns in Cr3 shift dramatically when using the full theory instead of simpler approximations. This shows capturing subtle spin interactions is key to understanding real magnetic materials. (Source: arXiv:2605.31203)
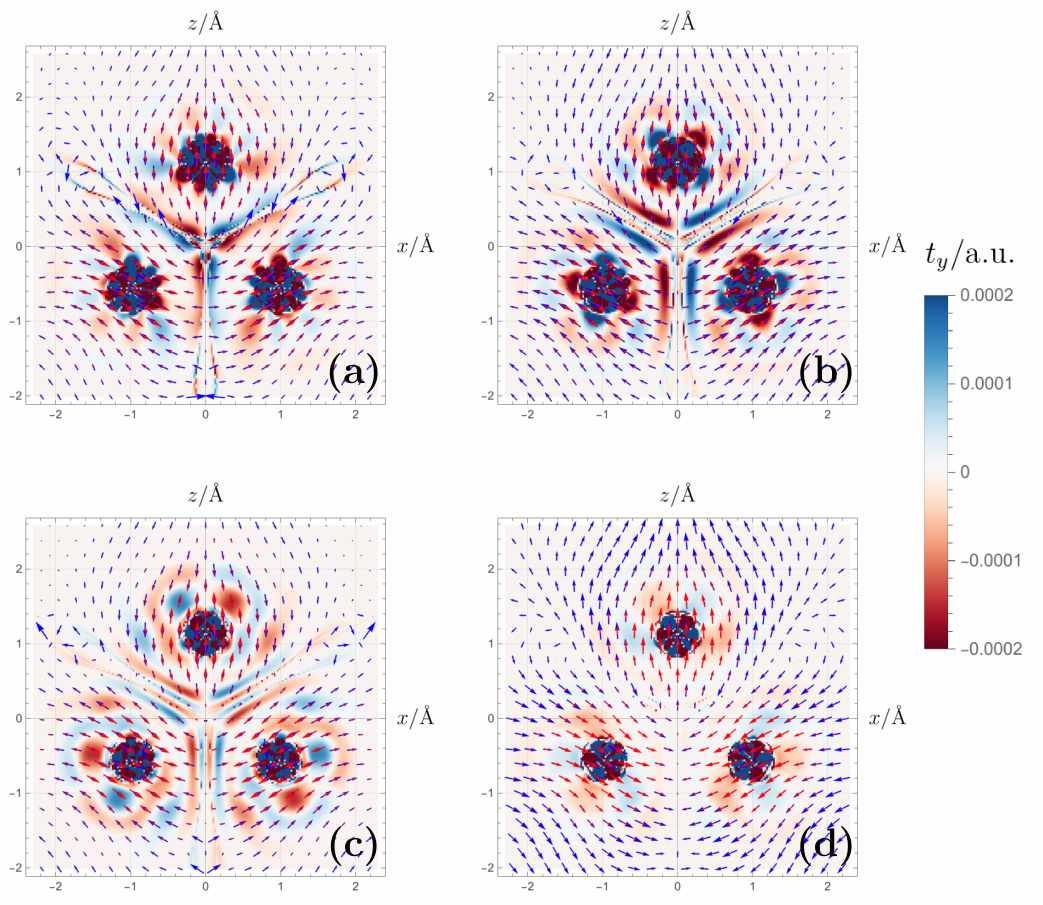
Magnetic torque patterns in Cr3 change dramatically with the choice of density functional. This sensitivity determines how well simulations capture real magnetic behavior in molecules and materials. (Source: arXiv:2605.31203)
But the method is not limited to semilocal functionals. Gaul argues that the same SU(2)‑rotation strategy extends to fully nonlocal functionals — hybrid functionals, for instance, that mix exact exchange — and, importantly, to the computation of relativistic response properties. To demonstrate, he calculates the hyperfine coupling constants in the high‑spin ground states of atomic uranium and the uranium ion, systems where spin‑orbit coupling is so enormous that a noncollinear treatment is mandatory. The hyperfine interaction, which depends on the spin density right at the nucleus, is a notoriously delicate observable. The fact that the NC‑SU(2) approach provides numerically stable results, while automatically satisfying the torque equations, is a strong hint that the method captures the essential physics without artificial constraints.
There is something philosophically satisfying about seeing the collinear and noncollinear worlds reconciled so cleanly. For years, the two camps in computational magnetism operated almost as separate subfields, each with its own codes, its own favourite functionals, and a vague sense that bridging them would be either impossible or hopelessly messy. Gaul’s paper quietly announces that the bridge is already there, hidden in the SU(2) structure that electrons have always obeyed. What felt like an awkward patch — the Scalmani–Frisch correction — was just the visible tip of a much deeper gauge invariance. That is the kind of insight that resonates beyond density functional theory. It reminds us that when a theoretical framework appears to fragment into mutually incompatible pieces, the fracture often reflects a choice of coordinates rather than a fundamental divide. The right rotation, the right conceptual dial, can make the fragmentation disappear.
The road ahead is clear. The NC‑SU(2) approach now needs to be implemented in mainstream electronic‑structure codes so that its predictions can be tested against experiment on real materials — frustrated magnets, heavy‑element compounds, spintronic devices — where collinear approximations are known to mislead. Gaul’s preprint provides the theoretical backbone; the next step is for the wider community to pick it up and see how far it can carry us. If history is any guide, a deep symmetry that connects seemingly disparate corners of a theory tends to unlock more than anyone initially expects. A simple rotation may turn out to have turned a page.
References
- Konstantin Gaul, Rigorous extension of semilocal collinear functionals to noncollinear DFT using SU(2) rotations, arXiv:2605.31203